無源植入性醫療器械是指植入人體后,不依賴外部能源(如電能、化學能)發揮其功能的醫療器械,如骨科植入物、心血管支架、人工關節等。這類產品與人體組織長期接觸,其生物相容性是決定其安全有效性的核心要素,也是技術審評的關注重點。與此隨著醫療器械產業的數字化轉型,信息系統集成服務在提升評價效率、確保數據完整性與可追溯性方面扮演著日益重要的角色。本文將系統闡述生物相容性評價的技術審評要求,并探討信息系統集成服務在此領域的應用價值及常見問題。
一、 生物相容性評價技術審評的核心要求
技術審評主要依據相關國家標準(如GB/T 16886系列)和國際標準(如ISO 10993系列),重點關注以下幾個方面:
- 評價策略的合理性:注冊申請人需根據產品的材料特性、與人體的接觸性質(如接觸部位、接觸時間)和接觸方式,制定科學、全面的生物學評價策略。審評關注策略是否覆蓋了所有潛在風險(如細胞毒性、致敏性、刺激或皮內反應、全身毒性、遺傳毒性、植入后局部反應等),以及豁免某些試驗的理由是否充分。
- 測試方法的合規性與科學性:試驗需在具有相應資質的實驗室進行,并遵循良好實驗室規范(GLP)。審評關注試驗設計(如樣品制備、浸提介質、浸提條件)、試驗方法(是否符合標準)、劑量選擇、對照設置等是否科學合理。對于創新材料或復雜器械,標準方法可能不適用,需提供方法學驗證資料。
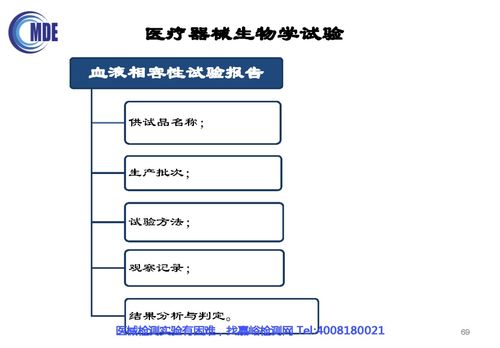
- 結果數據的充分性與可接受性:所有試驗結果均應清晰、完整、可追溯。審評關注結果是否支持產品生物相容性的安全結論,對任何陽性或可疑結果必須有合理的解釋和充分的風險控制措施(如材料純化工藝改進、清洗驗證等)。
- 風險-受益的綜合分析:生物相容性評價是風險管理的一部分。審評要求申請人將生物學試驗結果與產品的臨床受益相結合,進行綜合風險評估,確保產品的整體安全性在可接受范圍內。
二、 信息系統集成服務在評價中的應用價值
在生物相容性評價的復雜流程中,信息系統集成服務通過整合不同的軟件、硬件和數據資源,能夠顯著提升工作效率與合規水平:
- 數據全生命周期管理:集成實驗室信息管理系統(LIMS)、電子實驗記錄本(ELN)等,實現從試驗方案設計、原始數據采集、過程監控、結果分析到報告生成的全流程電子化、規范化管理,確保數據真實、完整、不可篡改且易于審計。
- 流程自動化與協同:自動化的工作流引擎可以規范評價流程,減少人為差錯,并促進研發、測試、質量、注冊各部門之間的高效協同。例如,系統可自動觸發樣品送檢、跟蹤試驗進度、提醒報告審核節點。
- 知識庫與決策支持:集成材料數據庫、歷史測試數據庫、法規標準庫,為評價策略的制定提供數據支持和參考,輔助進行更科學的評估與決策。
- 審評資料高效準備:系統可自動匯總、整理所有相關的試驗數據、報告和佐證材料,生成符合監管提交要求的電子資料包(eSTAR格式或類似),極大提高注冊申報資料的準備效率和質量。
三、 常見問題分析與建議
在技術審評及信息系統應用過程中,常見以下問題:
- 評價策略不完整或依據不足:常見于未充分評估長期植入產品的慢性毒性、致癌性等終點,或對材料可瀝濾物分析不足。建議:深入理解產品特性與相關標準,進行全面的風險識別,必要時咨詢有經驗的毒理學專家。
- 試驗設計或執行存在缺陷:如浸提比例不合理、浸提條件不能代表最壞臨床情況、陰性/陽性對照設置不當等。建議:加強與測試實驗室的溝通,確保試驗方案設計嚴謹,并嚴格監控試驗過程。
- 數據管理與追溯性差:傳統紙質記錄或孤立的電子表格難以滿足數據完整性(ALCOA+原則)要求。建議:引入或升級專業的信息管理系統(如LIMS),建立統一的數據管理平臺,實現全流程電子化追溯。
- 信息系統“孤島”與集成失效:不同部門或環節使用的系統互不聯通,數據需手動搬運,易出錯且效率低下。建議:在規劃之初就進行頂層設計,選擇開放性好、支持標準接口的系統,或委托專業的集成服務商,實現系統間的無縫對接與數據流暢通。
- 忽視系統驗證與合規:用于合規活動的信息系統本身需要經過嚴格的驗證,以確保其符合GxP規范。建議:按照相關指南(如GAMP 5)對信息系統進行完整的驗證(IQ/OQ/PQ),并保留完整的驗證文檔。
結論:無源植入性醫療器械的生物相容性評價是一項嚴謹的系統工程。企業不僅需要深入掌握技術審評的法規與科學要求,構建完善的評價體系,還應積極擁抱數字化變革,借助專業的信息系統集成服務,實現評價過程的智能化、規范化和高效化管理,從而從根本上提升產品質量與注冊成功率,保障患者安全。